Retinitis Pigmentoza (Gece Körlüğü)
Halk arasında Tavuk Karası Hastalığı ya da Gece Körlüğü olarak bilinen hastalık retina tabakasının en sık görülen distrofisidir. Retinitis pigmentosa (RP), fotoreseptör hücre ölümü ve retinal pigment epitel atrofisinin olduğu, sonuçta iki taraflı körlüğe neden olan genetik olarak heterojen bir retinopatidir. RP'nin yaygınlığı dünya çapında 1/7000 ila 1/3000 arasında değişmektedir. RP her yaşta başlayabilir. Genellikle 10 yaş civarında başlar ve görme bozukluğu 40-50 yaşlarında belirginleşir. Genel olarak erken başlangıçlı RP tipleri hızlı ilerleme eğilimindedir.
Retinitis pigmentozanın tipleri nelerdir?
RP’nin iki alt tipi vardır: 1)Non-sendromik Retinitis Pigmentoza ve 2) Sendromik Retinitis Pigmentoza . Non-sendromik RP; neden olan genin konumu ve ekspresyon özelliğine göre üç kategoriye ayrılır: otozomal dominant RP ( %15-25), otozom resesif RP (%5-20) ve X'e bağlı RP ( %10–15). Geri kalanların %40-50'si farklı fenotipik özelliklere sahiptir; bi-genetik RP veya mitokondriyal kalıtsal RP gibi ve nadirdir. Genel olarak, X'e bağlı RP'li hastalar, otozomal resesif RP'li hastalara göre daha ciddi hastalık fenotipleri sergilerken, otozomal dominant RP'li hastalar santral görmenin korunmasıyla en iyi prognoza sahip olanıdır.
Sendromik RP; RP hastalarının yaklaşık %20-%30’dur ve yaklaşık 30 farklı türü vardır, ancak genel olarak doğuştan metabolizma hataları ve siliyopatiler ile birlikte olan RP olarak 2’ye ayrılabilir. Doğuştan metabolizma hataları, metabolik yollardan birindeki (örneğin karbonhidrat, protein veya glikojen depolama yolları) önemli bir enzimin fonksiyonunun kaybolduğu geniş bir grup genetik bozukluğu içerir. Bu grup hastalıkların klinik olarak beyine yönelik bir tercihi vardır ve merkezi sinir sisteminin bir parçası olduğu için retinayı da etkileyebilir. Örnekler arasında yetişkin Refsum hastalığı (RP, nörodejenerasyon, ataksi, işitme kaybı, anozmi ve kalp/iskelet/deri tutulumu), Bassen-Kornzweig sendromu (RP, yağ malabsorbsiyonu, akanthasitoz, düşük kan kolesterolü, nörodejenerasyon) ve PHARC sendromu (polinöropati, işitme kaybı, ataksi, RP ve katarakt), Abetolipoproteinemi (spinoserebellar ataksi, retinal pigmenter dejenerasyon), nöronal seroid lipofuscinozis (demans, nöbet, pigmenter retinopati ile karakterize ciddi görme kaybı), mukopolisakkaridozlardan Hurler, Scheie, Sanflippo sendromunda (iskelet anomalileri, kalp problemleri, beyin ödemi, pigmenter retinopati) RP görülür.
Siliyopatiler, primer siliaların oluşumunu veya işlevini etkileyen bir grup hastalıktır. Silia, plazma zarının mikrotübüler uzantılarıdır ve neredeyse her hücre tipinin bir bileşenidir. Bu nedenle silialardaki genetik kusurlar tipik olarak birden fazla sistemi etkiler. Retinada, fotoreseptörlerin dış bölümleri silyumlar aracılığıyla iç bölümlerine bağlanır. Siliyopatilerde sıklıkla etkilenen diğer organlar iç kulak, böbrek, karaciğer ve merkezi sinir sistemidir. Retinal dejenerasyonla birlikte olduğu bilinen siliopatiler arasında Usher sendromu, Joubert sendromu (retina dejenerasyonu, zihinsel engellilik, polidaktili, ataksi), Senior-Loken Sendromu (retina dejenerasyonu ve nefronofitizis) ve Bardet-Biedl sendromu (RP, zihinsel engellilik, polidaktili, obezite ve hipogonadizm) yer alır. RP hastalarının %10’unda işitme kaybı izlenir. İşitme kaybı ve RP ile birlikte seyreden sendromlar Usher, Waardenburg sendromu, Alport sendromu ve Refsum hastalığıdır.
Retinitis Pigmentoza hastalarında semptomlar nasıl gelişir?
Retina dejenerasyonunun ilk bulguları fotoreseptör hücre stresinin başlaması ve dış segmentlerinin kısalmasının başlamasıyla kendini gösterir. İlk klinik belirti, gece görüşünde azalmadır. RP’li hastalar hastalığın erken dönemlerinde düşük ışık koşullarında veya ışıklı ortamlardan loş ortamlara hızlı adaptasyon gerektiren durumlarda iyi göremediklerini ifade ederler. Bazı kişiler, karşıdan gelen farların ve ortamdaki diğer parlak ışık kaynaklarından sonra karanlığa alışmalarının zorlaşması nedeniyle geceleri araç kullanmakta zorlandıklarını ifade ederler.
Hastalık ilerledikçe, santral retinada kon hücrelerinin dış segmentlerinin ilerleyici kaybı ve retinanın periferik bölgesindeki rod fotoreseptörlerin kaybıyla birlikte, görme alanı daralır ve görme keskinliği azalır. RP hastalarının çoğu yaşamlarının dördüncü on yılında yasal olarak kör olacak olsa da, hala bir miktar makula fonksiyonu kaldığından ışık görme kaybolmaz.
Klinik tanı bulguları nelerdir?
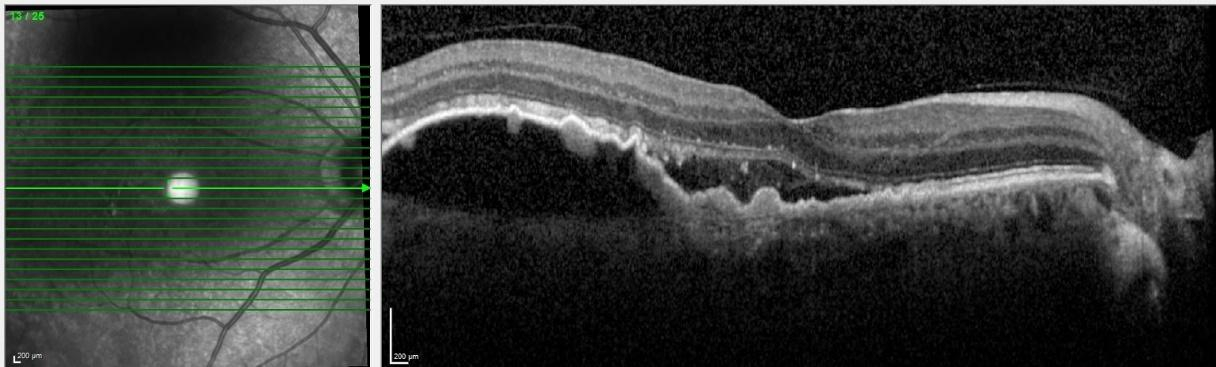
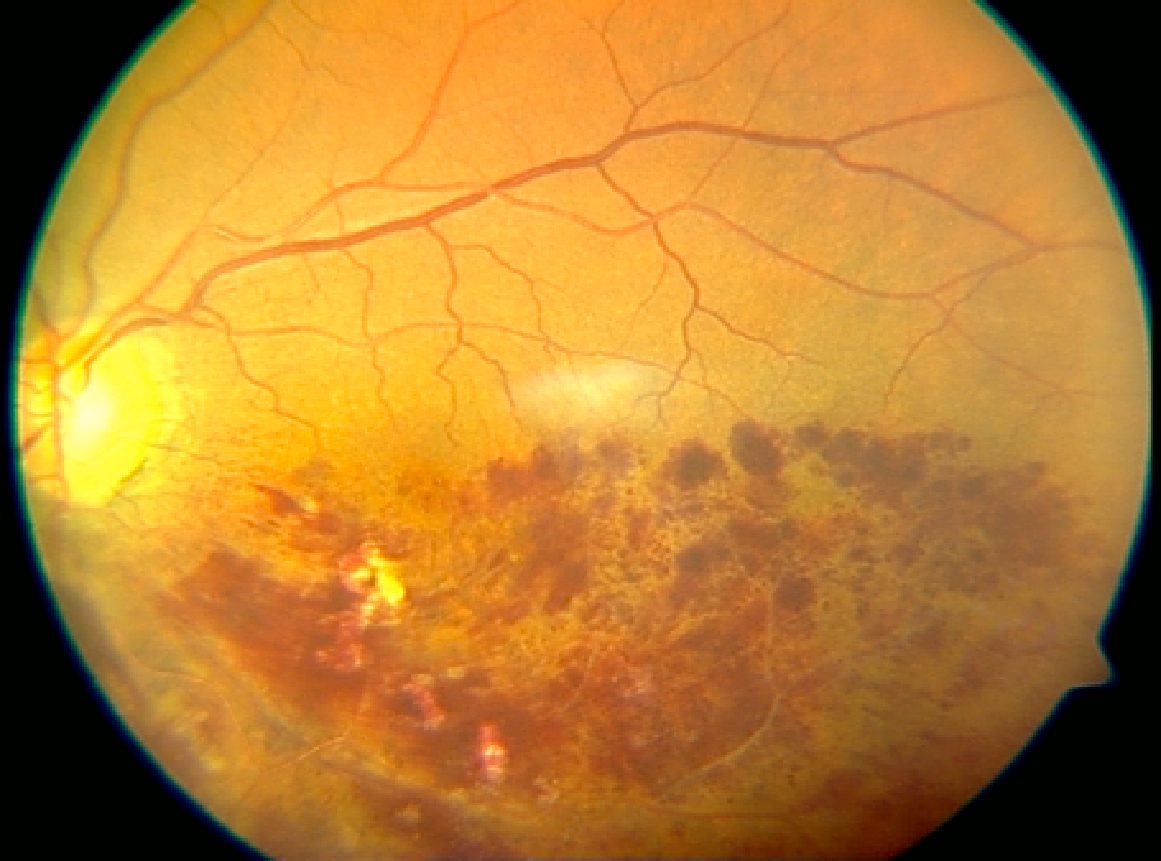
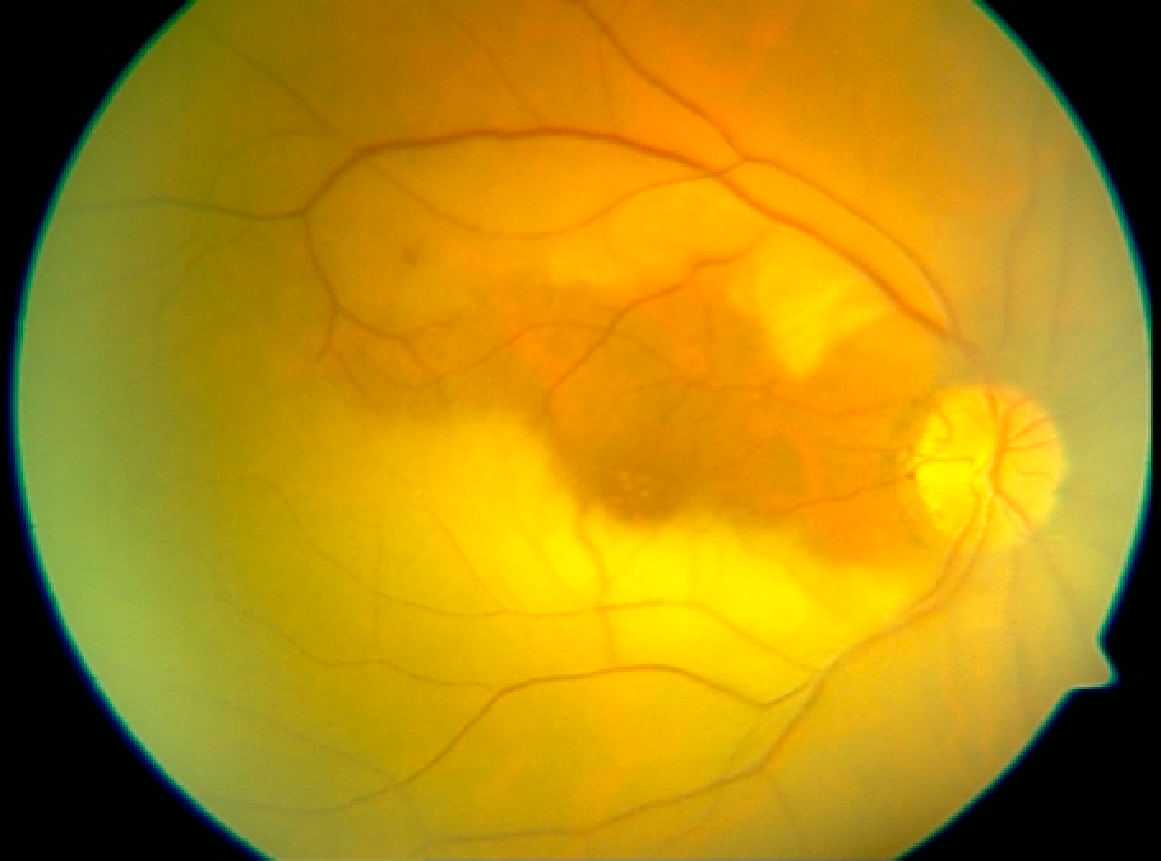
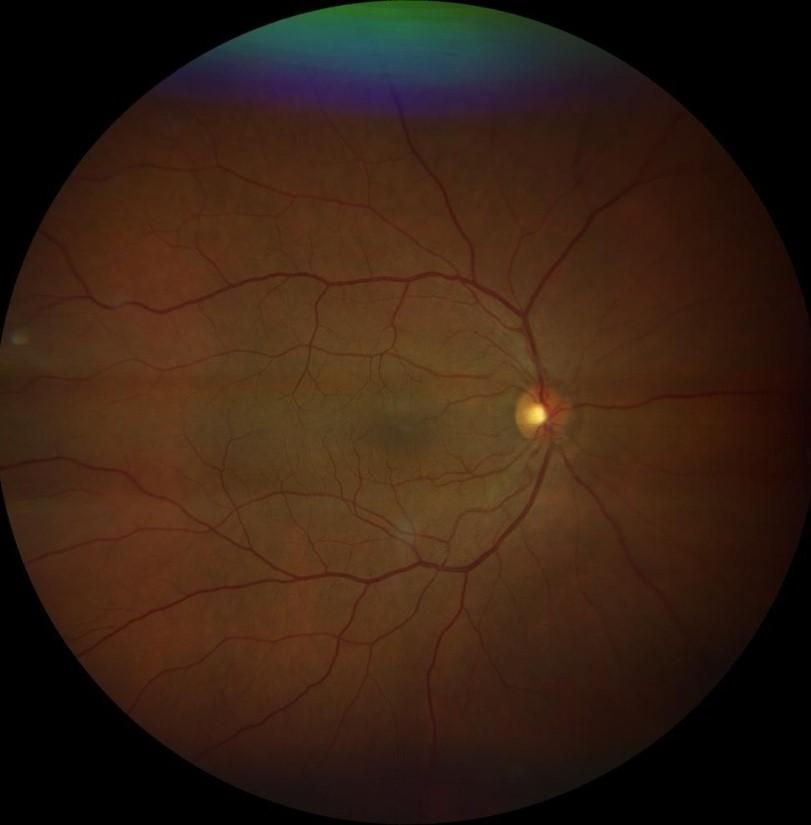
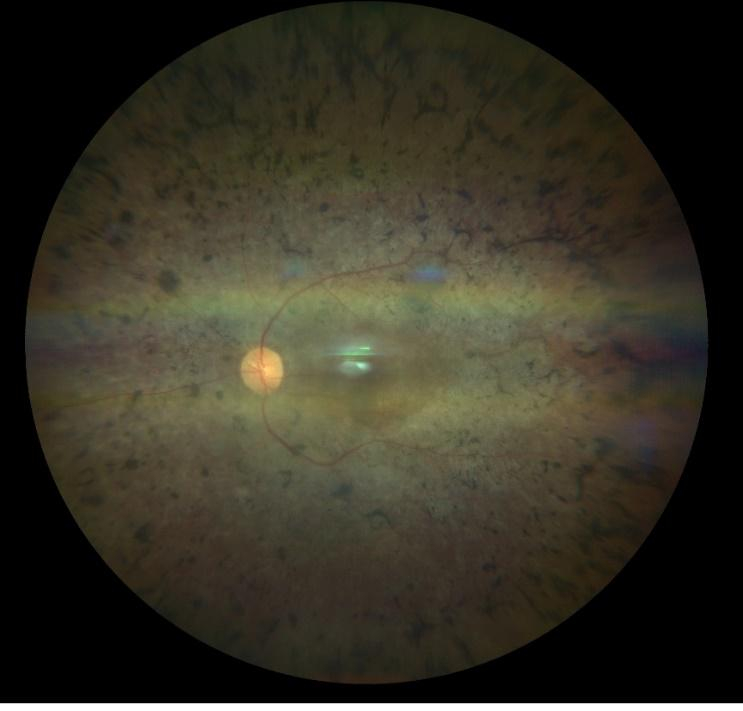
Göz bebekleri büyütülüp retina incelendiğinde RP’nin tipik üç klinik bulgusu vardır: 1) kemik spikülü şeklinde olan pigmentasyonu, 2) retinal damarlarda daralma ve 3) optik sinir solukluğu. Karakteristik kemik spikülü şeklinde olan melanin pigment birikintileri, ayrılıp retinadaki perivasküler bölgelere göç eden retina pigment epitel hücrelerinden kaynaklanmaktadır. Bu göçün kesin nedeni ve retinal damarların daralması tam olarak anlaşılamamıştır, ancak bunun çok sayıda fotoreseptörün ölümü nedeniyle azalan metabolik talepten kaynaklandığı düşünülmektedir.
Panretinal distrofi aşamasında gözlenen yaygın belirtiler arasında optik sinir başı druseni, kistoid maküla ödemi, vitreus hücreleri, epiretinal membranlar ve arka subkapsüler katarakt yer alır.
Resim: Retinitis pigmentozalı bir hastanın göz dibi görünümü.
Retinitis Pigmentoza hastalarında hastalık nasıl yönetilir?
Şu anda RP'li hastaların çoğu için iyileştirici bir tedavi mevcut değildir. RPE65 gen mutasyonuna sahip RP hastaları gen terapisini almaya uygundur, bunlarda RP’li hastaların küçük bir kısmını oluşturmaktadır. RP'li hastaların çoğu, A vitamini takviyeleri, güneş ışığından korunma, görsel yardımlar, tıbbi ve cerrahi müdahaleler dahil olmak üzere geleneksel tedavi seçenekleri ile tedavi edilmektedir. Bu müdahaleler semptomları yönetmeyi, oftalmik komplikasyonları önlemeyi ve hastalığın ilerlemesini yavaşlatmayı amaçlar ancak hastalığı iyileştirmez.
Erken müdahalenin hastalığın ilerlemesini yavaşlattığı genel olarak kabul edilmektedir. Bu nedenle, ailesel genetik bilgi ve belirlenmiş fenotipik özelliklerle birlikte erken moleküler tanı, etkili müdahale için gereklidir.
Son yıllarda RP'nin genetik nedenleri daha iyi anlaşılmakta ve hastalıkla mücadeleye yönelik yeni tedaviler geliştirilmektedir.
Retinitis pigmentosa için gelişmiş tedavi yöntemleri. ( a ) Hastalığa neden olan geni değiştirmek için terapötik genin virüs aracılı enjeksiyonunu kullanan gen tedavisi; ( b ) Dejenere hücreleri değiştirmek ve kalan retina nöronlarıyla sinaptik bağlantılar oluşturmak için kök hücrelerin enjekte edildiği hücre tedavisi; ( c ) Koni fonksiyonunu geri kazandırmak için dejenerasyona uğramış retinaya fotosensitif proteinlerin sokulduğu optogenetik ve ( d ) Hastalığın özellikle erken evrelerinde ek tedavi olarak kullanılan nörotrofik faktörler, anti-apoptotik ajanlar ve antioksidanlar gibi nöroprotektif ajanlar.
Gene özgü veya mutasyona özgü araştırmalar, fotoreseptörlerde normal gen ifadesini yeniden sağlamak amacı taşır.
Diğer araştırmalar, retinayı fonksiyonel fotoreseptörlerle yeniden doldurmak için retina progenitör hücrelerinin (veya oküler olmayan kök hücrelerin) göze nakledilmesini içeren hücre replasman tedavisini içerir. Embriyonik kök hücreleri, uyarılmış pluripotent kök hücreler ve mezenkimal kök hücreler gözdeki hedef konumlara iletilerek fotoreseptörlerin ölümünü yavaşlatır veya önler. Kök hücrelerin kullanıldığı klinik araştırmalar nispeten güvenli bir profil oluşturmuş olsa da, uzun vadeli etkinlik, reddedilme ve inflamasyon dahil olmak üzere birçok engel ve endişe devam etmektedir.
Nöroprotektif ajan tedavisi, iyi tolere edilen ve az yan etkisi olan, en eski ve en yaygın kullanılan yaklaşımlardan biridir. Genellikle hastalığın erken evrelerinde kullanılır ve diğer evrelerde yardımcı tedavi olarak da kullanılabilir. Nöroprotektif ajanlar temel olarak nörotrofik faktörleri, anti-apoptotik ajanları ve antioksidanları içerir. Çalışmalardan 10 mg/kg/günden DHA (Omega 3) alımının ERG fonksiyon kaybını azalttığı gösterilmiştir. Ayrıca DHA takviyesinin 12 yaş altı hastalarda rod ERG fonksiyonunun korunmasında, 12 yaş ve üzeri hastalarda ise kon ERG fonksiyonunun korunmasında faydalı olduğu bilinmektedir. Yapılan pek çok çalışma ile 15000 IU/gün A vitamininin veya 15000 IU/gün A vitamini+ 3 IU/gün E vitamininin RP de progresyonu yavaşlattığını, 12 mg/gün lutein takviyesinin, A Vitamini alan retinitis pigmentozalı sigara içmeyen yetişkinlerde mid-periferik görme alanı kaybını yavaşlattığını bilmekteyiz.
Son yıllarda nöroprotektif amaçla retinanın dejeneratif hastalıklarında uygulanan bir diğer yöntem ekzosom uygulamalarıdır. Hücreler tarafından salgılanan küçük veziküller olan ekzosomlar, biyolojik olarak aktif molekülleri belirli hücrelere ve dokulara iletme kabiliyetleri nedeniyle son zamanlarda RP için potansiyel bir tedavi seçeneği olarak dikkat çekmiştir. Ekzosomlar, hücreler tarafından salgılanan ve tüm biyolojik sıvılarda bulunan bir tür hücre dışı veziküldür. Bağışıklık tepkisi, kardiyovasküler hastalıklar, merkezi sinir sistemi hastalıkları ve kanser dahil olmak üzere çeşitli fizyolojik ve patolojik süreçlerde rol oynarlar. Bu nano boyutlu yapılar (çapı 30–100 nm) lipitler, nükleik asitler ve proteinler (yani sinyal proteinleri, metabolik enzimler ve antijenler) gibi çeşitli moleküller içerir. Eksozomların büyüme faktörlerinin ve diğer biyolojik olarak aktif moleküllerin alıcı hücrelere aktarılması da dahil olmak üzere çeşitli mekanizmalar aracılığıyla etki edebileceği düşünülmektedir. Bu, gen ifadesinde ve hücresel işlevde değişikliklere, bağışıklık tepkilerinin modülasyonuna ve apoptozisin baskılanmasına yol açarak fotoreseptör hücreleri koruyabilecek mekanizmaları tetikleyebileceği in vivo çalışmalarda gösterilmiştir. Ancak RP tedavisinde ekzosomların güvenliğini ve etkinliğini göstermek için daha sağlam klinik öncesi ve klinik verilere ihtiyaç vardır.
Elektriksel stimülasyonu (ES), hedef dokulara mikro akımın iletildiği farmakolojik olmayan bir tedavidir. Transkraniyal ES tedavisi, depresyon, otizm spektrum bozukluğu, inme, travmatik beyin hasarı, Alzheimer hastalığı ve Parkinson hastalığı gibi beyin hastalıklarında hem fonksiyonel hem de yapısal koruma sağladığı yıllardır bilinmektedir. 2002 yılında Morimoto ve ark. tarafından ES'nin gözde tedavi potansiyeli ilk kez rapor edilmiştir. ES, sıçan optik sinirinin kesilen ucuna uygulandı ve aksotomize edilmiş retina ganglion hücrelerinin hayatta kalma oranını arttırdı. ES, RP, yaşa bağlı makula dejenerasyonu, optik nöropati, glokom ve retinal arter tıkanıklığı olan hastaları tedavi etmek için uygulandı. Elektrotların konumu ile tanımlanan transkorneal, transpalpebral, transdermal ve transorbital yolla alternatif akım stimülasyonu yoluyla göze elektrik akımı verilebilmektedir. ES'nin nöronal apoptozu önlediği, nöronal rejenerasyonu desteklediği, Müller hücrelerinde nörotrofik faktör üretimini artırdığı, retinal mikroglial aktivasyonunu engellediği, retinal kan akışını artırdığı ve beyin plastisitesini modüle ettiği çalışmalarla desteklenmiştir.
RP’nin oftalmik komplikasyonlarının tedavisi görme keskinliğine olumlu katkıda bulunacaktır. Kistoid maküla ödeminin tedavisi, katarakt cerrahisi, epiretinal membran ya da makula deliklerinin cerrahi tedavisi RP hastalarının yaşam kalitesini arttırır.
Prognoz
Retinitis pigmentosa'nın görsel prognozu, hastalığın evresine, genetik faktörlere ve tedaviye yanıtına bağlı olarak değişir. Erken tanı ve müdahale, hastalığın ilerlemesini yavaşlatabilir ve yaşam kalitesini iyileştirebilir. Gelecekteki tedavi seçenekleri ve araştırmalar, RP'li bireyler için daha iyi prognozlar ve yaşam kalitesi iyileştirmeleri sunma potansiyeline sahiptir.